![]() Despite the fact that medical device malfunctions often lead to severe patient injury or even death, the U.S. Food and Drug Administration (FDA) is seeking to relax the adverse event reporting requirement, further slowing down an already slow process of recognizing device problems and potentially putting patients at greater risk.
Despite the fact that medical device malfunctions often lead to severe patient injury or even death, the U.S. Food and Drug Administration (FDA) is seeking to relax the adverse event reporting requirement, further slowing down an already slow process of recognizing device problems and potentially putting patients at greater risk.
The 2017 draft of the Medical Device User Fee Amendments, which is renegotiated every five years, would allow device manufacturers to report malfunctions to the U.S. Food and Drug Administration (FDA) quarterly instead of within the current 30-day requirement. The House has already reauthorized the new agreement, which now awaits Senate approval.
Who Currently Reports Device Malfunctions?
Currently, only device manufacturers and user facilities are required to notify the FDA of suspected medical device malfunctions leading to serious injuries and deaths. The Medical Device Guardians Act, legislation to require individual physicians to report adverse events, was introduced in the U.S. House in 2016, but the measure was not included into the 21st Century Cures Act. The failed measure sought to allow anyone who observes an adverse event – doctors, nurses, institutions, or patients – to be able to easily make the event public, available, and accessible.
GAO Says Adverse Events Underreported
In February 2017, a report issued by the Government Accountability Office stated that adverse event reports to the FDA are often underreported, incomplete, erroneous, and not timely. This is in agreement with a 2015 survey of reporting standards at 17 hospitals by the FDA, which concluded that the majority of the hospitals studied did not file timely reports of injuries and deaths due to malfunctioning medical devices. But despite the findings, the FDA did not impose penalties against hospitals that failed to comply with the reporting requirements.
Other Medical Devices News
The FDA warns that Synovo Total Hip Systems, implanted after 2019, pose risks of failure and injury. Patients experiencing symptoms should consult our CSS lawyers to explore legal options for compensation.
Thousands of hernia mesh products have been recalled, but qualifying for a claim requires proof that a defective hernia mesh implant caused your injury.
Damages from a defective hernia mesh implant vary based on each person’s injuries and circumstances. An experienced dangerous medical device attorney can help identify all the losses you may be entitled to recover.
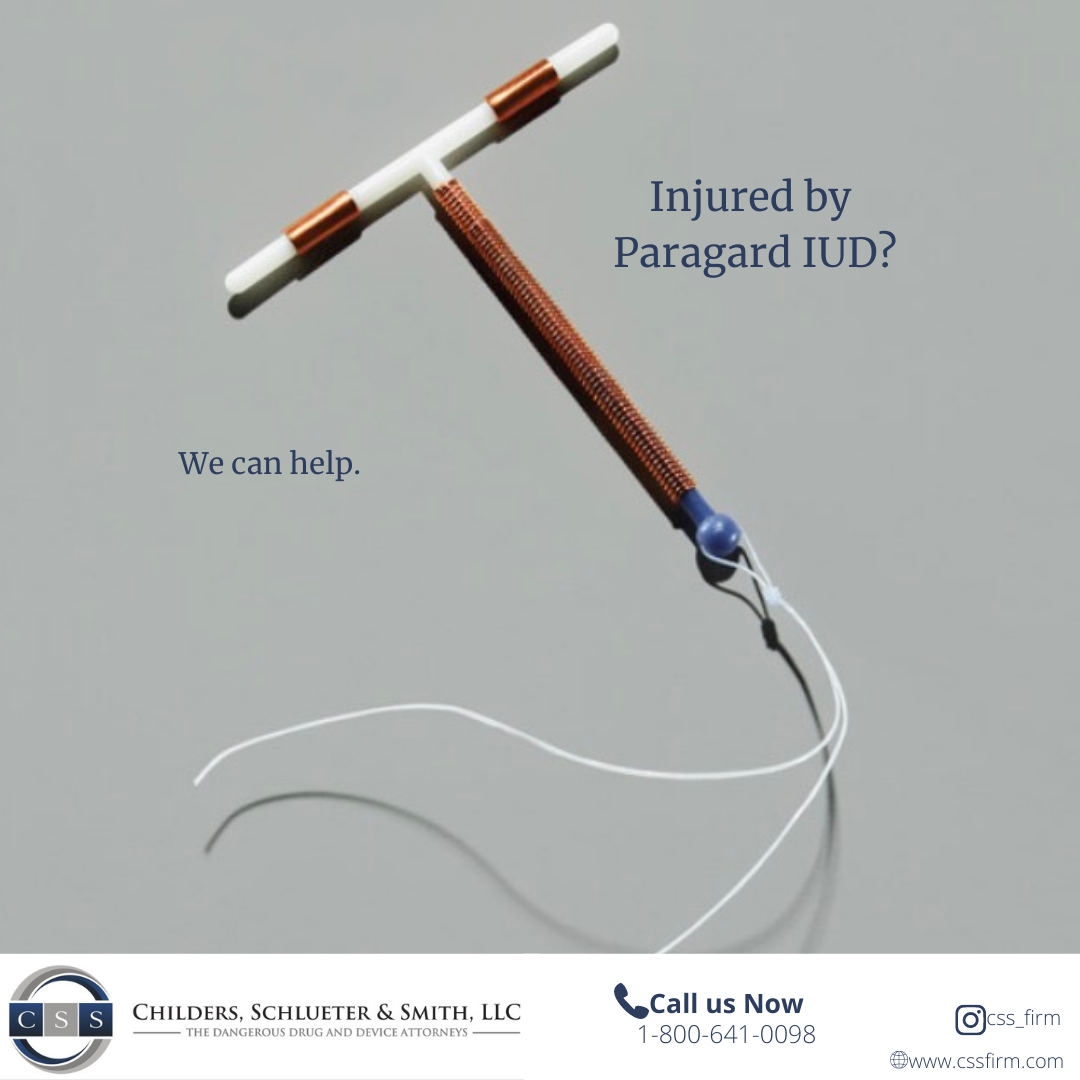
Women nationwide are filing lawsuits after suffering injuries from Paragard IUD breakage—alleging the manufacturer failed to warn of the device’s dangerous design defects.

A voluntary recall affects over 3.5 million Philips Respironics CPAP, BiPAP, and ventilator devices due to foam degradation risks, potentially endangering sleep apnea patients’ health and safety.
Philips Respironics has recalled certain CPAP, BiPAP, and ventilator devices due to degrading sound foam that may release harmful particles and chemicals, posing serious health risks to users.